转自:药渡

酪氨酸激酶2(TYK2)是一种非受体酪氨酸激酶,属于JAK家族,还包括JAK1, JAK2和JAK3。TYK2调节IL-23和IL-12受体下游的信号转导,是各种自身免疫性疾病的一个有吸引力的靶点。与目前批准的JAK抑制剂相比,选择性TYK2抑制提供了不同的临床特征。然而,TYK2对其他JAK家族成员的选择性很难通过抑制催化活性激酶结构域的小分子来实现。最近用Deucravacitinib成功地靶向TYK2假激酶结构域作为实现异构体选择性的策略。本文描述了针对假激酶结构域的选择性TYK2抑制剂的优化,从而发现了临床候选药物ABBV-712(21)。
已知JAK家族激酶(JAK1, JAK2, JAK3和TYK2)介导几种引起卵细胞炎症的细胞因子的信号传导。近年来,小分子JAK抑制剂已被证明在一系列免疫介导疾病中具有临床应用价值。批准的JAK1抑制剂结合在被称为Janus Homology 1(JH1)的结构域,从而通过阻断ATP进入该位点并阻止随后的磷酸化事件来阻止激酶的催化活性。这种抑制导致多种促炎介质(如IFN-α、IFN-γ、IL-6、IL-8、IL-15、IL-31等)下游的JAK-STAT通路被阻断。小分子泛JAK和JAK1选择性抑制剂在自身免疫性疾病(包括类风湿关节炎、溃疡性结肠炎、牛皮癣和特应性皮炎)中已显示出临床疗效。与JAK1抑制相比,TYK2的选择性抑制提供了不同的特征,其活性更局限于IL-23、IL-12和干扰素-α(IFN-α)途径。
有证据表明IL-12和IL-23在炎症和自身免疫性疾病病理中的作用。单克隆抗体Ustekinumab已证实IL-12/23通路的临床意义,该单克隆抗体靶向IL-12和IL-23的常见p40亚基,被批准用于治疗银屑病、银屑病关节炎、溃疡性结肠炎等。此外,靶向IL-23的p19亚基的单克隆抗体已被批准用于几种自身免疫性疾病。Risankizumab被批准用于治疗银屑病,银屑病关节炎,和克罗恩病,Guselkumab被批准用于治疗银屑病12和银屑病关节炎,13和Tildrakizumab用于治疗银屑病。因此,介导IL-12和IL-23受体下游信号传导的TYK2已成为小分子发现的靶标,因此发现了一系列候选化合物。
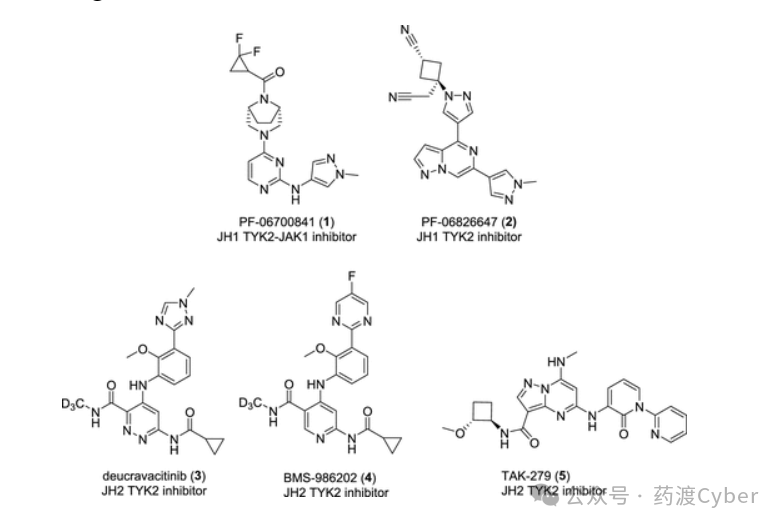
鉴于JAK家族激酶之间的激酶结构域具有高序列同源性,选择性ATP竞争性TYK2抑制剂的开发一直具有挑战性,很少有化合物进入临床。PF-06700841(1)是一种双重TYK2/JAK1抑制剂,目前正在研究用于多种自身免疫性适应症。
此外,一种更具选择性的TYK2抑制剂PF-06826647(2)正处于临床开发阶段,用于治疗银屑病、溃疡性结肠炎和化脓性汗腺炎。由于在TYK2和其他JAK家族成员之间的JH2结构域的显著差异,对TYK2抑制的变构方法显著提高了对其他JAK家族激酶的选择性。Deucravacitinib是一种选择性TYK2抑制剂,最近被批准用于牛皮癣,目前正在评估其他适应症,是第一个结合和稳定TYK2的JH2结构域并变构抑制蛋白激酶功能的TYK2临床候选药物。在寻找与Deucravacitinib结构不同的TYK2抑制剂的过程中,BMS-986202被确定并进入临床I期试验。武田还推进了一项变构TYK2药物TAK-279,最近报告了一项治疗牛皮癣的IIb期临床试验的正面数据。(图1)
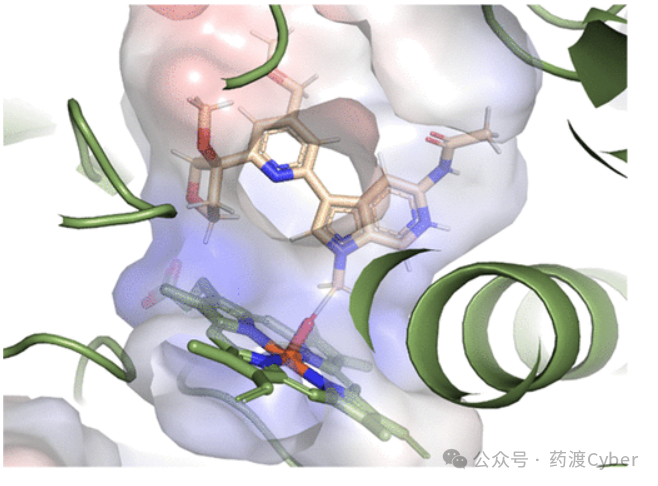
这篇文章对TYK2伪激酶-激酶结构域结构与结合的小分子2-氯-N-(2(环丙烷羧基)吡啶-4-基)苯酰胺(PDB 4OLI)进行了早期分析,特别对TYK2中JH2结合腔提供了独特见解(图2)。值得注意的是,吡啶酰胺片段结合在JH1和JH2结构域,这种双重结合可能由于高激酶结构域同源性而降低了JAK家族的选择性。因此,在之后专注于设计JH2结构域的特异性化合物。
现有一些已发表的JH2结构域结构(JAK1 PDB 4L01;JAK2 PDB 4FVP)将有助于确定多样性的关键区域,可以指导设计JH2结构选择性。当叠加这些结构时,出现了显著差异,包括TYK2中的Val640(JAK1中的Ile620和JAK2中的Leu579)和TYK2中的Val603(JAK1中的Ile597和JAK2中的Ile559)。这些细微的序列变化被推测为驱动异构体JH2口袋内不同的结合轮廓。在“pseudo-gatekeeper”基团位置观察到最丰富的氨基酸多样性:TYK2中的Thr687,JAK1中的Glu667和JAK2中的Gln626。这些不同的残基在该区域产生独特的静电环境和空间约束。具体来说,在TYK2中,与JAK1或JAK2相比,Lys642侧链在较小的TYK2“pseudo-gatekeeper”基团存在时具有更大的构象灵活性。
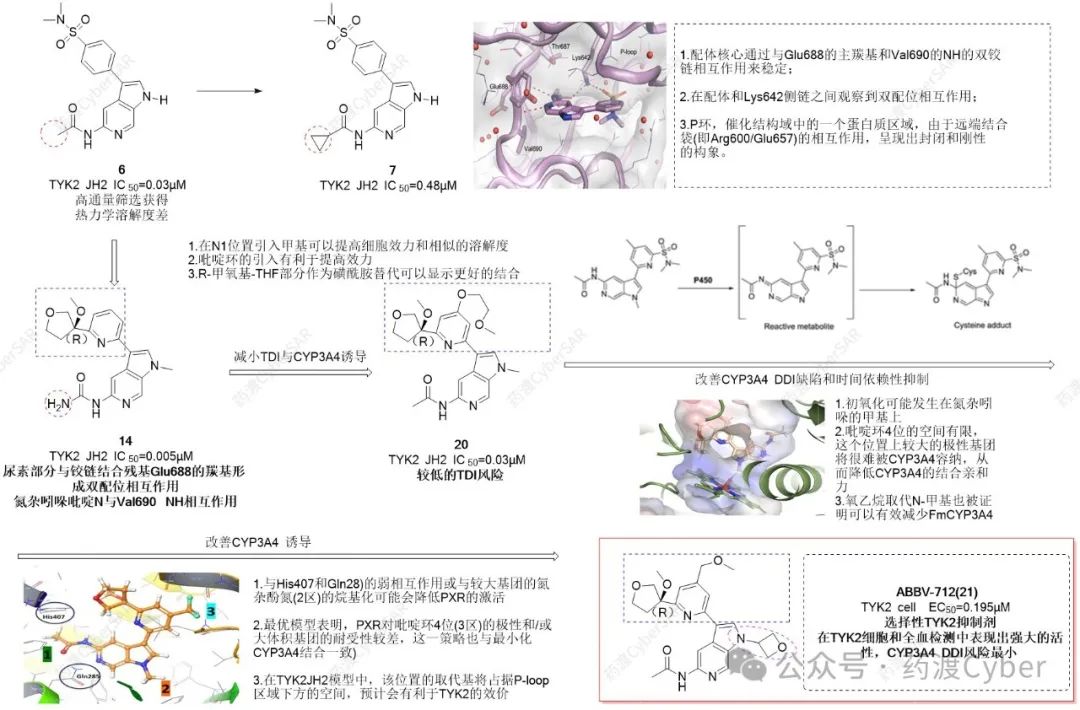
早期的Hit-To-Lead工作从TYK2 JH2稳定剂的高通量筛选中确定了6-氮杂吲哚支架。优化得到的磺胺6(表1)与TYK2 JH2结构域结合(IC50=0.03μM, TRFRET结合实验),对TYK2和JAK1 JH1结构域的特异性均>200倍(IC50分别=6和40μM, TRFRET结合实验)。在IL-12诱导的人T细胞p-STAT4实验中,观察到良好的TYK2细胞活性(96孔和384孔格式的EC50分别为0.12和0.32μM)。与结合选择性一致,没有观察到JAK1、JAK2和JAK3的细胞活性(分别通过IL-6诱导的人TF-1细胞中的p-STAT3、促红细胞生成素诱导的人UT-7细胞中的p-STAT5和IL-2诱导的人t-blast细胞中的p-STAT5测量,每个细胞的EC50>19μM)。磺胺6是了解JAK家族选择性的有效工具,然而,它的热力学溶解度很差(在pH 7.4的磷酸盐缓冲液中为1.2μM)。随后获得了与环丙酰胺7密切相关的晶体结构(图3),以支持设计和优化工作(PDB 8TB5)。
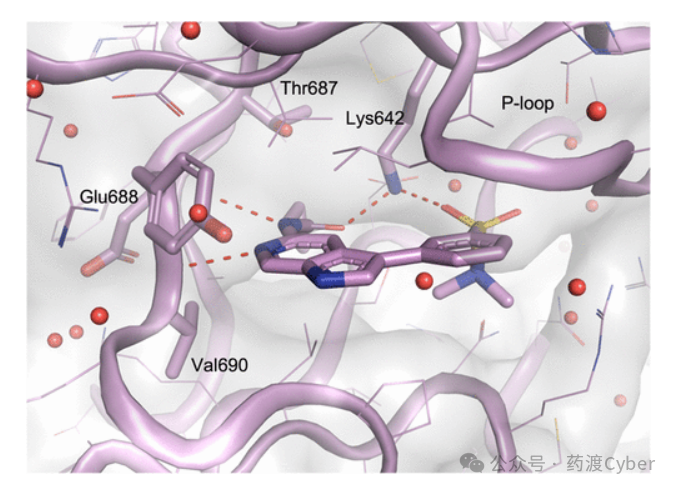
表1. TYK2选择性及理化性质的优化

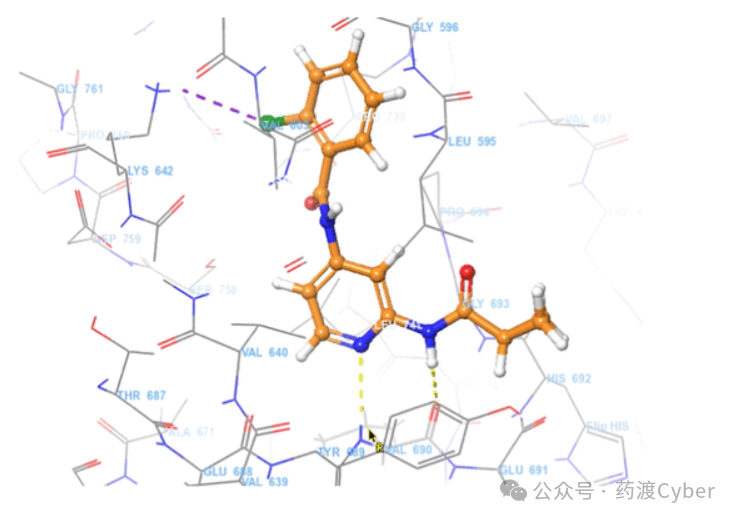
通过7的X射线结构,可以确定配体设计的关键区域(图3)。首先,配体核心通过与Glu688的主羰基和Val690的NH的双铰链相互作用来稳定。由此产生的配体取向将环丙基投射到“pseudo-gatekeeper”Thr687后面的一个结合口袋中,这与蛋白质主链产生了轻微的冲突,推测这与6相比导致活性下降。其次,在配体和Lys642侧链之间观察到双配位相互作用。酰胺羰基和一个磺酰胺氧都参与了与两个赖氨酸N-Hs的双重结合。这种与Lys642的相互作用被假设提高了对其他JAK亚型的结合亲和力和选择性,这是由于侧链相对于“pseudo-gatekeeper”的定位。第三,P环,催化结构域中的一个蛋白质区域,由于远端结合袋(即Arg600/Glu657)的相互作用,呈现出封闭和刚性的构象。这三个区域是迭代设计过程中的重点领域。
环丙基酰胺7的共晶结构为了解配体与TYK2的JH2结构域之间的相互作用提供了有价值的见解。然而,环丙酰胺7的效力低于乙酰胺6,因此选择6作为进一步优化的起点,旨在提高效力和可成药性质。
与化合物6相比,在N1位置(8)引入甲基可以提高细胞效力和相似的溶解度。X射线结构分析表明,双环偶氮唑与苯基环(θ=1.1°)接近共平面。扭角分析证实,这种构象的不利因素约为2kcal/mol(图4)。相比之下,如果2-吡啶环具有类似的共面构象,则计算出的不利要小得多(例如,化合物9,图4)。事实上,吡啶9在结合实验中几乎是3倍的效力,但令人惊讶的是在细胞实验中是同等效力的。然而,9显示出显著降低的非结合清除率,几乎低了一个单位的c logP值,并被选为进一步优化的起点。基于结合亲和力,乙基砜在10中作为磺酰胺替代品耐受性良好,但在细胞测定中也显示出显著的效力损失。试图用氧乙烷取代砜乙基以降低c logP(11)导致进一步的效价损失。由于铅类似物9(2.5μM)的溶解度较低,研究人员探索了硅法来识别具有改善溶解度潜力的其他砜/磺胺替代品。分子动力学(MD)模拟使用甲氧基-THF部分作为磺酰胺替代(12),突出了在整个模拟过程中观察到的THF氧、酰胺羰基和Lys642之间独特、稳定的双配位相互作用(图5)。相比之下,化合物12的对映体13没有显示出稳定的双配位相互作用(MD结果未显示)。其中14具有良好的结合亲和力(IC50=5nM)和细胞效力(EC50=60nM)。X射线晶体结构证实THF环氧确实指向Lys642,尿素羰基与Lys642相互作用。尿素部分与铰链结合残基Glu688的羰基形成双配位相互作用,氮杂吲哚吡啶N与Val690 NH相互作用(图6)。
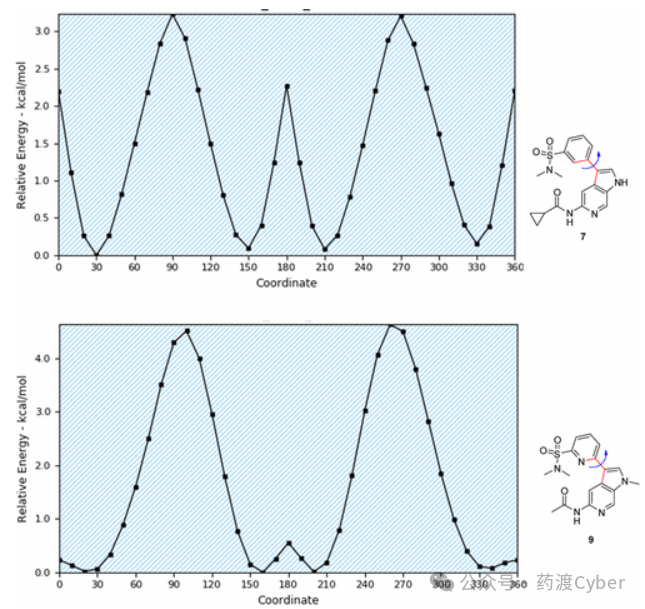
9的相对能量示意(上图:化合物7中氮唑核心与苯基之间1.1°的扭转角约为2kcal/mol;下图:化合物9中氮唑核与2-吡啶环之间的扭转角为1.1°,相对能量明显降低)

图5.在TYK2 JH2的分子动力学模拟中,THF氧和酰胺羰基与Lys642的双配位相互作用以及与Val690的铰链相互作用(化合物12)

图6.结合TYK2 JH2结构域的化合物14的晶体结构显示了THF环氧和尿素羰基与Lys642(PDB 8TB6)的双配位相互作用。
虽然14的效能是令人鼓舞的,但该化合物的被动渗透性很差,考虑到化合物中有多个氢键供体,这也许不足为奇。尿素被酰胺部分取代,如在12和13中,导致效力降低。化合物13的效价最弱,可能是由于THF氧的取向远离Lys642,这与MD结果一致。虽然THF氧在14的X射线中的位置不能明确指定,但观察到THF立体异构体(例如,12和13)之间的效力一致变化。为了确定正确的立体化学性质,采用振动圆二色性(VCD)对常见中间体2-溴-6-(3-甲氧基四氢呋喃-3-基)吡啶(方案1,步骤g)的对映体结构进行了确定。(S)-2-溴-6-(3-甲氧基四氢呋喃-3-基)吡啶用于制备活性较低的对映体13,证实了活性更高的(R)对映体12中的THF氧原子与Lys642形成氢键。含有4-吡啶基取代基的化合物占据p环下的空间(表2),显示出更高的效力,但也存在不良的药物-药物相互作用风险。例如,腈15和16表现出明显的CYP3A4 DDI牺牲潜力(>60% FmCYP3A4), -CHF2, -OCH3和-OCHF2类似物17,18和19表现出CYP3A4的时间依赖性抑制(在50μM下变化20%)和CYP3A4诱导(2倍)。乙醚20仍有明显的CYP3A4诱导作用,但TDI风险降低。由于这些多重DDI挑战,随后的努力集中在更全面的方法来设计化合物来解决上述CYP3A4 DDI缺陷。

方案1.化合物12和13的合成
表2.减小TDI与CYP3A4诱导的SARS研究

CYP3A4 DDI缺陷和时间依赖性抑制
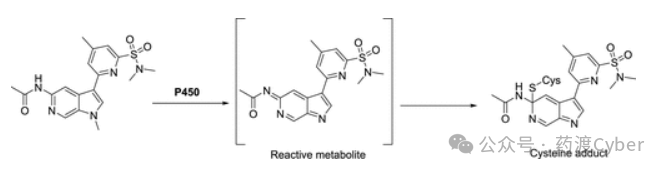
假设CYP3A4的时间依赖性抑制是N-去甲基化随后氧化的结果,导致形成可能与CYP3A4中的半胱氨酸发生反应的代谢物(图7)。替代甲基以减缓初始去甲基化的选择被认为是一种初步方法。然而,由于CYP3A4介导的代谢在这种化学型中很常见,并且CYP3A4的缺陷也被确定为一个常见的问题,实验人员决定转而专注于优化减少与CYP3A4的结合,作为并行解决这两个问题的途径。使用Molecular Forecaster impact软件,生成了该化学型的结合假设(图8)。根据该模型,初氧化可能发生在氮杂吲哚的甲基上。引人注目的是,吡啶环4位的空间有限,假设在这个位置上较大的极性基团将很难被CYP3A4容纳,从而降低CYP3A4的结合亲和力。例如,化合物20、22和23具有较低的CYP3A4代谢比例,与较小的基团如16和19相比,对CYP3A4的亲和力降低,支持之前的假设。用氧乙烷取代N-甲基在18和21中也被证明可以有效减少FmCYP3A4。此外,还考虑了这种4-吡啶基取代对CYP3A4诱导的影响。
CYP3A4诱导
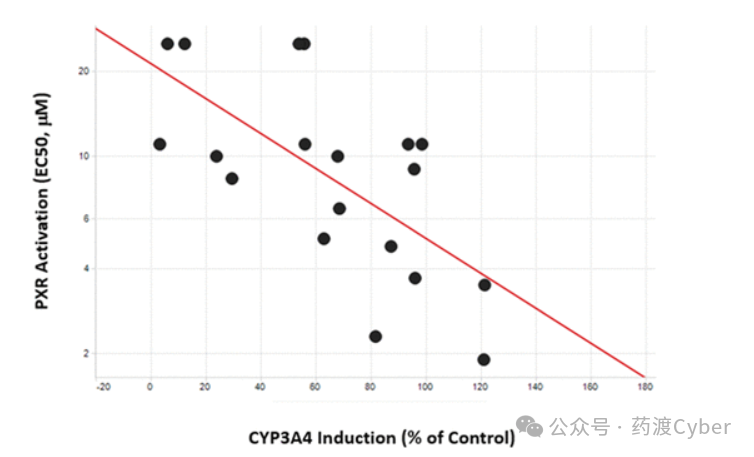
CYP3A4的诱导通常与核受体妊娠素X受体(PXR)的激活有关。为了证实PXR实际上对化学型特异性CYP3A4诱导负责,在PXR激活试验中评估了19种氮杂吲哚。观察到PXR激活(EC50)与CYP3A4诱导(%,图9)之间存在合理的相关性(R2=0.49),表明PXR激活的缓解会降低CYP3A4诱导。利用PXR的配体结合域(LBD)的已知X射线结构5A86、4XHD、3R8D和1NRL,生成了氮杂吲哚系列的潜在结合模式(图10)。
2=0.49)
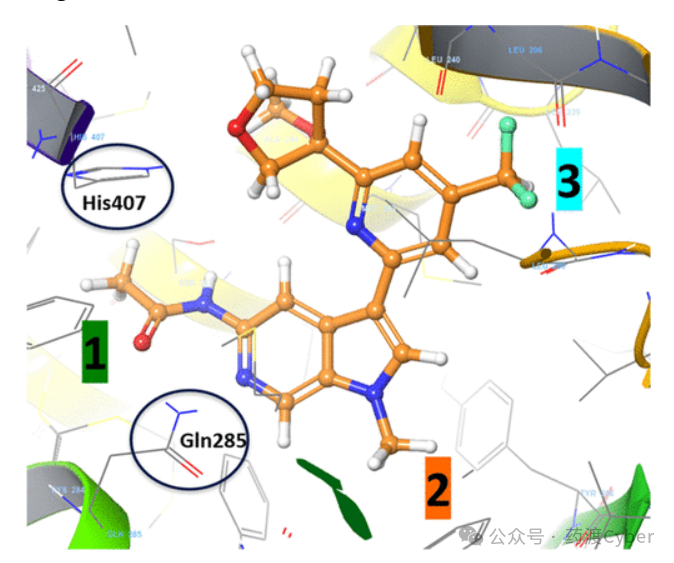
对所提出结合模式的分析表明,与His407和Gln285(图10中的1区)的弱相互作用或与较大基团的氮杂酚氮(2区)的烷基化可能会降低PXR的激活。最优模型表明,PXR对吡啶环4位(3区)的极性和/或大体积基团的耐受性较差,这一策略也与最小化CYP3A4结合一致(见上)。在TYK2 JH2模型中,该位置的取代基将占据P-loop区域下方的空间,预计会有利于TYK2的效价。基于这些集体结构评价,设计了吡啶环4位取代基的关键类似物,包括化合物21、22和23(表3)。
表3.选定化合物的体外谱图

令人欣慰的是,这三种化合物对CYP3A4代谢的依赖程度降低,CYP3A4代谢的比例较低,从而降低了它们的不利因素。21和23的时间依赖性抑制潜力略高于预期,此外,这三种类似物对CYP3A4的诱导潜力相对较低。基于CYP3A4 TDI和诱导在可接受范围内的事实,得出化合物21-23具有低成药性风险的结论。最后,成功地证明了这三个化合物对TYK2的细胞效力,提高了溶解度(>100μM),并在细胞分析中对其他JAK家族成员(>20μM)具有良好的选择性,证实了之前的假设,即通过靶向TYK2的假激酶结构域更容易实现选择性。
大鼠PK
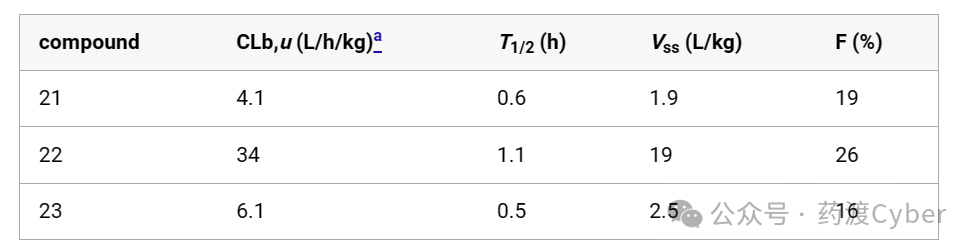
建立对体内药代动力学的早期理解对于优化化合物进入探索性毒性研究的进展至关重要。多参数优化有助于在低剂量大鼠PK研究中鉴定出几种类似物(表4)。化合物21显示出较低的体内非结合清除率(4.1L/h/kg)。尽管半衰期(0.6h)和口服生物利用度(19%)较低,化合物21仍可用于进一步的体内研究。仲醇22在TYK2细胞效能和最小CYP3A4 DDI负荷方面表现出类似的理想平衡,但表现出明显更高的非结合血液清除率(34L/h/kg),因此不太理想。从仲醇转换为伯醇(23)导致较低的未结合清除率(6L/h/kg)和较低的口服生物利用度(16%)。鉴于这种有希望的体内PK谱与优化的TYK2效价、溶解度和最小化的CYP3A4依赖性相平衡,21和23在犬和猴的PK研究中进一步进行了分析。
表4.大鼠体内药代动力学特性(按1mg/kg口服和静脉给药)
21和23的哺乳动物PK
化合物21和23的药代动力学在多个物种中进行了评估,以便计算预测的人类PK参数以进一步确定优先级(表5)。化合物21在犬和猴子中显示低至中等的非结合清除率(分别为0.46和2.3L/h/kg)。犬的半衰期比猴子长(4.5h比1.2h),这是由于犬的游离清除率特别低。口服给药对猴子的暴露率和生物利用度均为中等(F=17%),对犬的暴露率和生物利用度均为高(F=88%)。与化合物21相比,化合物23在犬和猴体内具有更高的未结合清除率(分别为2.5和4.7L/h/kg)。
表5.化合物21和23在全血中药动学性质的测定
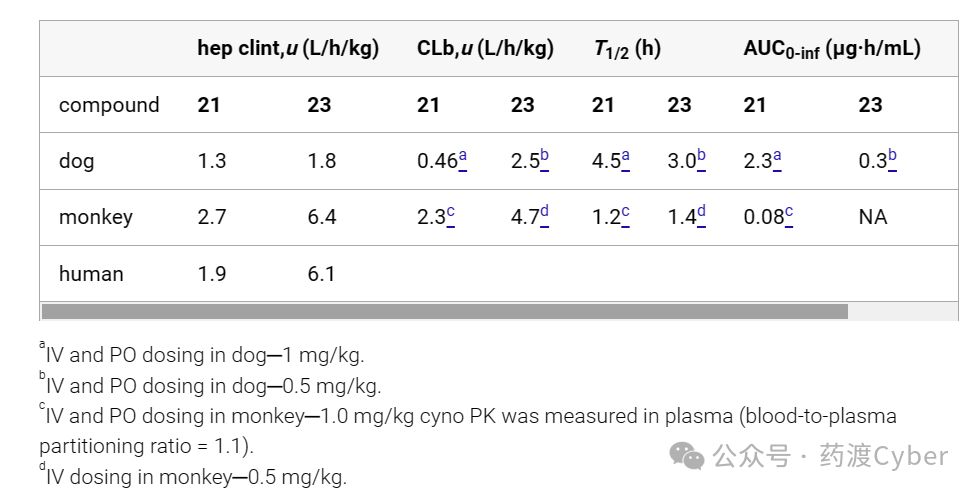
表6.化合物21的人类PK预测
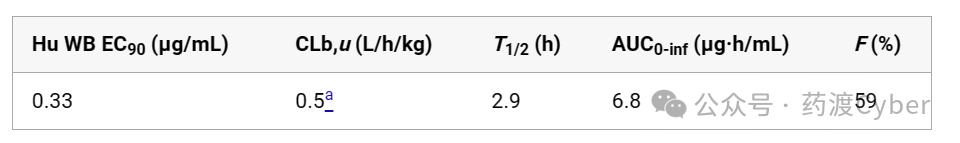
基于较好的非结合清除率值,选择化合物21进行进一步分析。初步计算了化合物21的人体PK参数。根据肝细胞清除率数据(表6),采用体外体内外推法(IVIVE)计算人体预测清除率(0.5L/h/kg)。以人TYK2全血EC90值(0.33μg/mL)作为Css的靶点计算预测人类有效AUC(6.8μg/mL*h)。EC90值是为了通过抑制该途径获得最大的疗效,尽管保持Css>EC50有望提供可测量的疗效反应。利用这些参数,计算出人体初始有效剂量为350mg。
化合物21的体内研究
评价化合物21的药效学作用对小鼠体内模型的影响。首先,通过IL-12刺激小鼠全血,测定STAT4磷酸化水平(EC50=1.4μM,0.63μg/mL),证实TYK2在小鼠体内的活性。尽管与人相比,化合物21的效价降低了8倍(EC50=0.16μM,0.07575μg/mL),但可接受的小鼠PK使化合物21能够在体内研究中达到足够的暴露已覆盖靶标。在TYK2抑制的急性PD模型中,小鼠口服剂量分别为10、30、100、300和600mg/kg,并在给药后30分钟(预期Cmax时间点)注射IL-12/IL-18。4小时后评估血清样本的IFN-γ水平。化合物21在100,300和600mg/kg剂量下具有剂量依赖性,可抑制IFN-γ的产生(图11),其最大暴露量显著高于小鼠全血EC50。
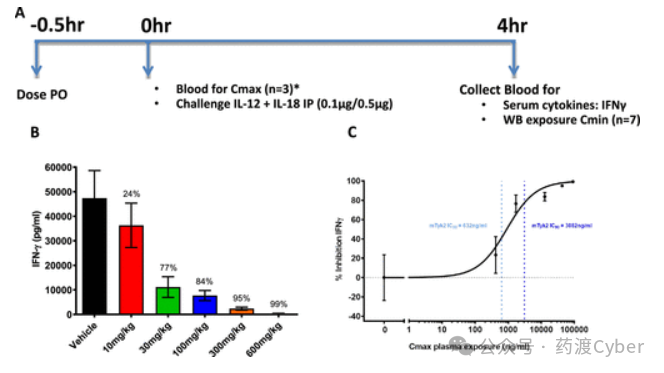
max暴露反应
采用小鼠IL-23微环诱导的耳部皮炎模型来评估Leys等人描述的化合物21的体内疗效。单次尾静脉注射IL-23微环(MC)可触发强烈的皮肤病理,激活IL-23/IL-17通路,导致第7天耳厚显著增加。在整个研究过程中,测量耳朵厚度从基线变化的百分比(图12A)。在整个研究过程中,假手术小鼠的耳朵厚度没有变化,而接受治疗的动物(第7-11天)的耳朵厚度呈剂量依赖性减少,到第11天,100mg/kg的化合物21给药BID或QD的抑制作用达到58-61%。
相比之下,选择抗IL-23 p40抗体作为机制阳性对照来评估该模型,在第7天和第9天以30mg/kg BID给药后,耳部厚度减少了60%。所有测试剂量的最大药物暴露水平均高于TYK2体外全血EC50(图12C)。
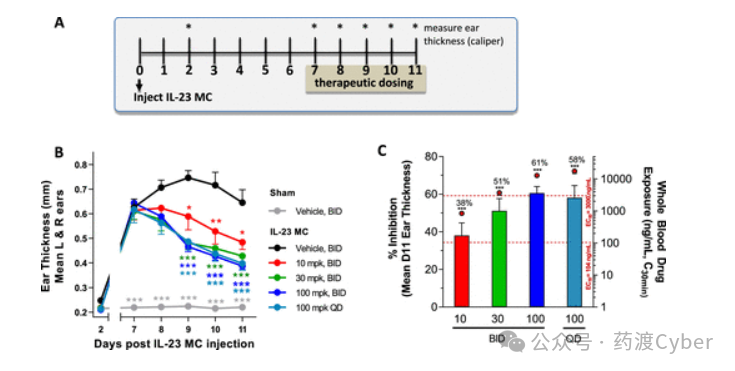
图12.小鼠IL-23诱导的耳部皮炎模型:(A)研究设计;(B)化合物21对耳部厚度的剂量依赖性降低;(C)化合物21的疗效与Cmax暴露反应
结论
化合物21是一种选择性TYK2抑制剂,是通过对一系列结合并稳定TYK2假激酶(JH2)结构域的氮杂吲哚进行多参数优化而发现的。在化合物14的X射线结构的指导下,优化TYK2的细胞和全血效力,结合药物样性质鉴定出非常有效和选择性的化合物;然而,许多化合物受到CYP3A4 TDI或CYP3A4诱导。通过CYP3A4活性位点的计算建模实现了对CYP3A4 TDI的缓解。通过确定PXR激活与CYP3A4诱导之间的良好相关性,解决了CYP3A4诱导问题;SAR优化以PXR结合位点的设计为指导。化合物21在TYK2细胞和全血检测中表现出强大的活性,CYP3A4 DDI风险最小。由于预测的人体清除率较低,并且基于人TYK2全血EC90靶向Css的有效AUC进行了调整,因此获得了350mg的预测有效剂量。最终,化合物21被指定为临床候选化合物ABBV-712。
文章来源
J. Med. Chem. 2023, 66, 14335−14356

1.药渡CyberSAR结合药物设计思想,挖掘了文献及专利报道的活性的结构,通过CyberSAR以方便快速获得研发人员兴趣靶向结构,以供开拓思路,就本文涉及Tyrosine-protein kinase TYK2(Homo sapiens)举例如下:
2.在靶点界面选中“化学空间”选项标签下级联“聚类结构视图”选项卡,可以将CyberSAR平台收录的文献和专利具有关于靶点相关实验测试活性的分子以“母核结构聚类”的形式展示。其中“绿色字体高亮的”为文献报道的体外酶、细胞活性测试实验中IC50<100nM的活性分子结构、具体实验、实验结果及实验来源。
3.在靶点界面选中“化学空间”选项标签下级联“原始结构视图”选项卡,可以将CyberSAR平台收录的文献具有关于靶点相关实验测试活性的分子以“研发阶段时光轴“的形式展示。其中绿色高亮展示了潜力Hit。
4.在靶点界面选中“适应症”选项标签,可以可视化直观分析现收集的各类大数据。
5.在靶点界面选中“实验设计”选项标签,可以将CyberSAR平台收录的文献相关靶点相关实验设计进行展示,为实验设计提供思路与支持。